Supercapacitors
Another one of our research focuses on modeling of novel nanostructured materials for electrochemical double layer capacitors (EDLC) or supercapacitors using room temperature ionic liquids (RTILs) as electrolyte. Our MD simulations using simplistic (coarse-grained) and fully atomistic force fields have studied EDL structure near flat and corrugated electrode surfaces as well as in nanopores. These studies showed that details of structural reorganization inside the EDL profoundly affect the energetics of adsorptive storage and ultimately the viability of this technology for practical applications. The latter is illustrated by our recent study of RTIL on graphite surfaces showing that slight differences in surface roughness qualitatively change the dependence and values of electrode capacitance and can noticeably increase the energy storage of the capacitor.
Our atomistic MD simulations of RTIL electrolytes in nanostructured electrodes also provided a significant insight into how the interplay between ion size/chemical structure and dimensions of nanopores in the electrode can result in non-trivial dependence of the energy storage by the capacitor as a function of applied voltage and electrode nanostructure. For example, simulations of EMIM/TFSI RTIL in slit nanopores allowed us to investigate the influence of ionic packing in charged nanopores on the EDL capacitance and obtain a fundamental molecular level understanding governing some intriguing experimental observations, such as e.g. the "volcano" effect. The latter refers to a sharp increase of electrode capacitance as dimensions of the nanopore become comparable to ion sizes.
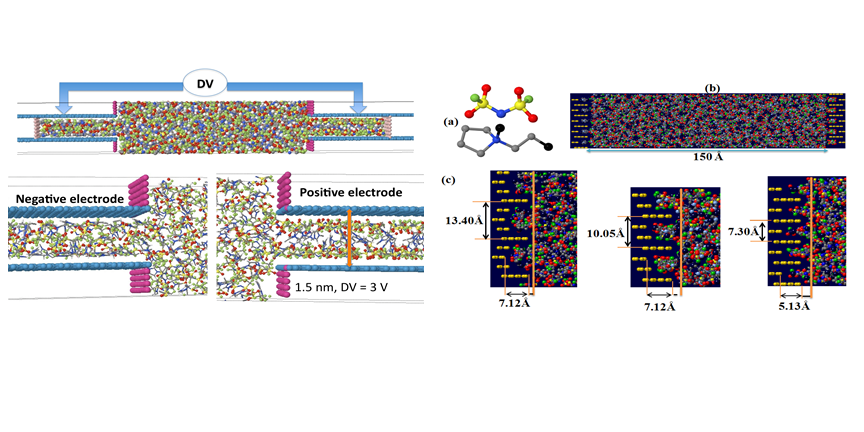
One of the important insights obtained from our simulations is that both for nanoporous electrodes and for flat electrodes with nanopatterned surfaces, the capacitance of electrodes is not symmetric and depends on the match/mismatch of ions chemical structure with electrode nanostructure. Such dependence potentially opens a tremendous opportunity to tune electrode and counterion chemical structures to maximize the performance of individual electrodes independently. Utilization of the state-of-the-art methodology which allows to model these systems at constant electrode potential allows us to provide unprecedented insight into influence of molecular level details on perfomance of supercapacitos. In tight collaboration with experimentalists we are currently working on optimizing the electrode structure and electrolyte compositions to significantly improve energy storage density in these devices.